cBioPortal - the Fred Hutch instance
Edit this Page via GitHub Comment by Filing an Issue Have Questions? Ask them here.Introduction to the Fred Hutch instance of cBioPortal and a demonstration of its usage in the context of research data.
Learning Objectives
After reading this article, you will learn:
- What cBioPortal is and how it can help with your research.
- What the Fred Hutch instance of cBioPortal is and steps to take to upload your data onto the instance.
- Ways to format your data for upload into cBioPortal.
What is cBioportal?
- cBioPortal is a free web-based tool that provides access to a variety of published cancer genomic datasets.
- Designed with ease of use in mind, cBioPortal is suitable for researchers without specialized knowledge in bioinformatics.
- It supports a wide range of data types including genetic, transcriptomic, proteomic and clinical information, allowing for a thorough analysis in one place.
- It allows users to visually explore patterns and changes across different cancers via its interactive tools.
- The platform integrates clinical information with genetic data, helping researchers explore this interaction easily.
What can you do with cBioportal?
There are innumerable ways you can leverage the vast amount of data on cBioPortal. In the example below, we demonstrate how you can harness some of the capabilities of cBioPortal to facilitate your own research.
Note: The examples below are demonstrated using the public instance of cBioPortal but can be replicated in the Fred Hutch instance as well)
As an example, let us say you identified KRAS as an important gene in cancer. Let’s see how we can use cBioPortal to expand this observation leveraging publicly available datasets.
Question 1: How often is KRAS mutated in cancers?
-
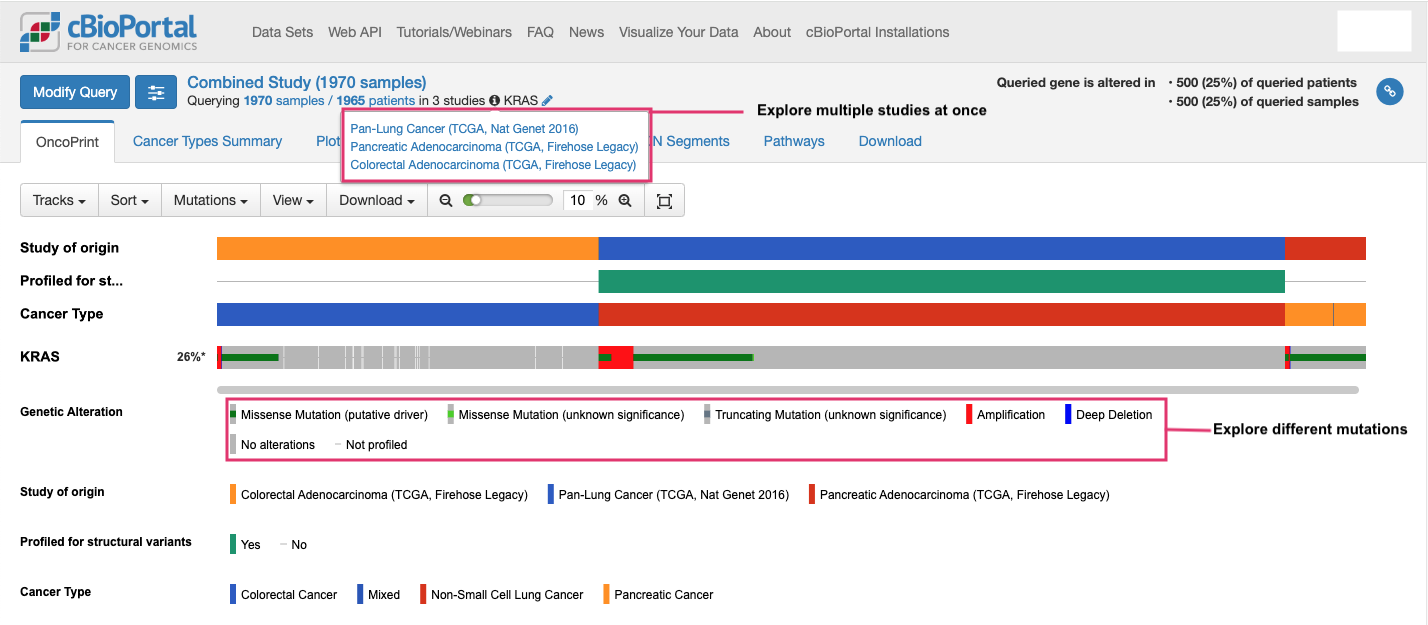
Using cBioPortal, you can explore the frequency of KRAS mutations in different cancers, for example here we look at colorectal, lung, and pancreatic cancers. This analysis shows that KRAS is mutated in 26% of patients across these studies. You can similarly investigate other studies (over 400 publicly available datasets) on cBioPortal.
-
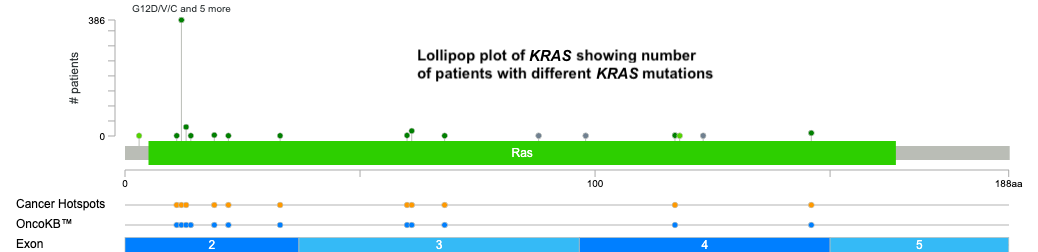
With cBioPortal, you can explore the different types of mutations in KRAS, such as single-nucleotide variations, insertions, deletions, and copy number changes. These can be visualized in OncoPrint format like above (showing mutations in genes for each sample), lollipop format like below (showing where mutations occur on the protein), and many others.
-
Additionally, cBioPortal’s direct integration with external databases such as ClinVar, COSMIC, etc. allows you to evaluate the impact of these genomic alterations. Here you can see each mutation in KRAS and its prediction of pathogenicity by OncoKB and COSMIC databases.
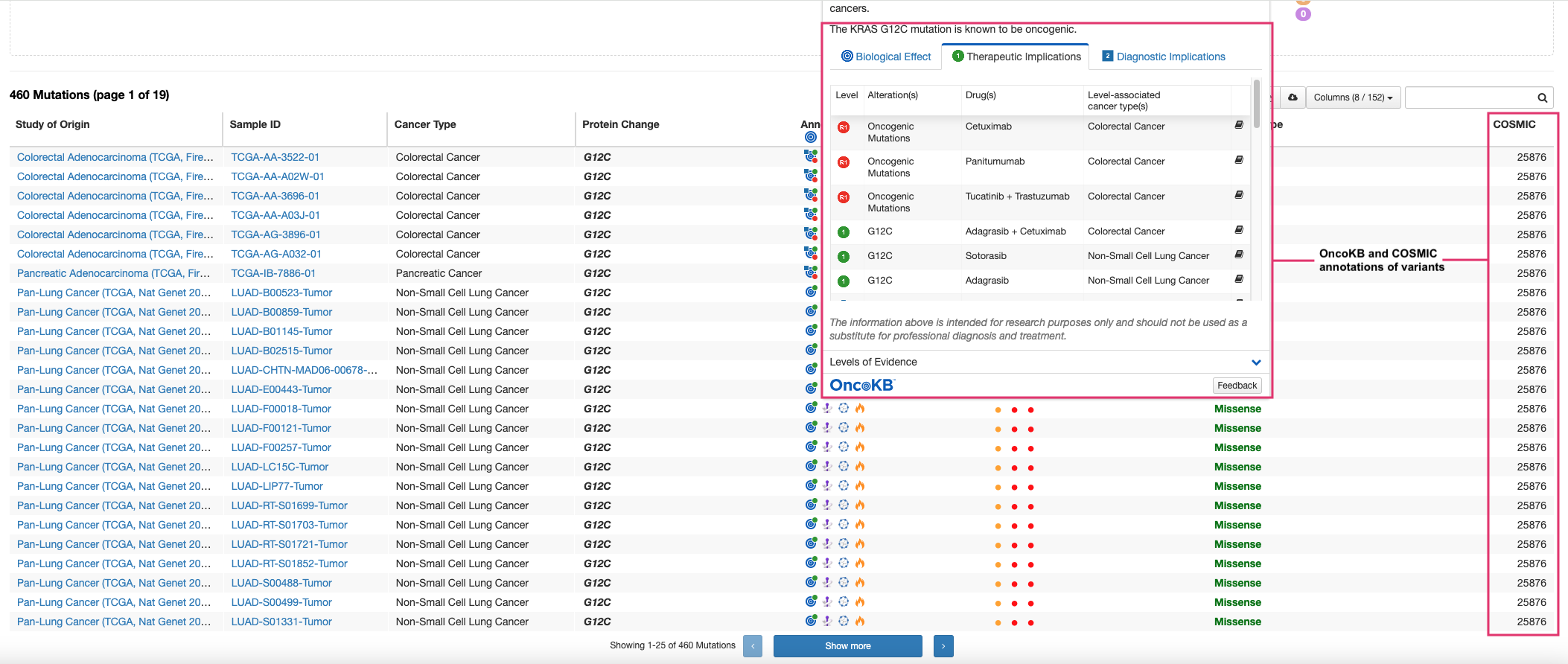
Thus with cBioPortal, exploring published multi-omics data becomes easier without needing to download raw data files, process them, and manually create visualizations. It also provides you with a one-stop shop to evaluate specific mutations by referencing external databases as well. The platform makes the entire process seamless and user-friendly.
Question 2: Are mutations in KRAS associated with any clinical parameters such as sex, age, etc.?
-
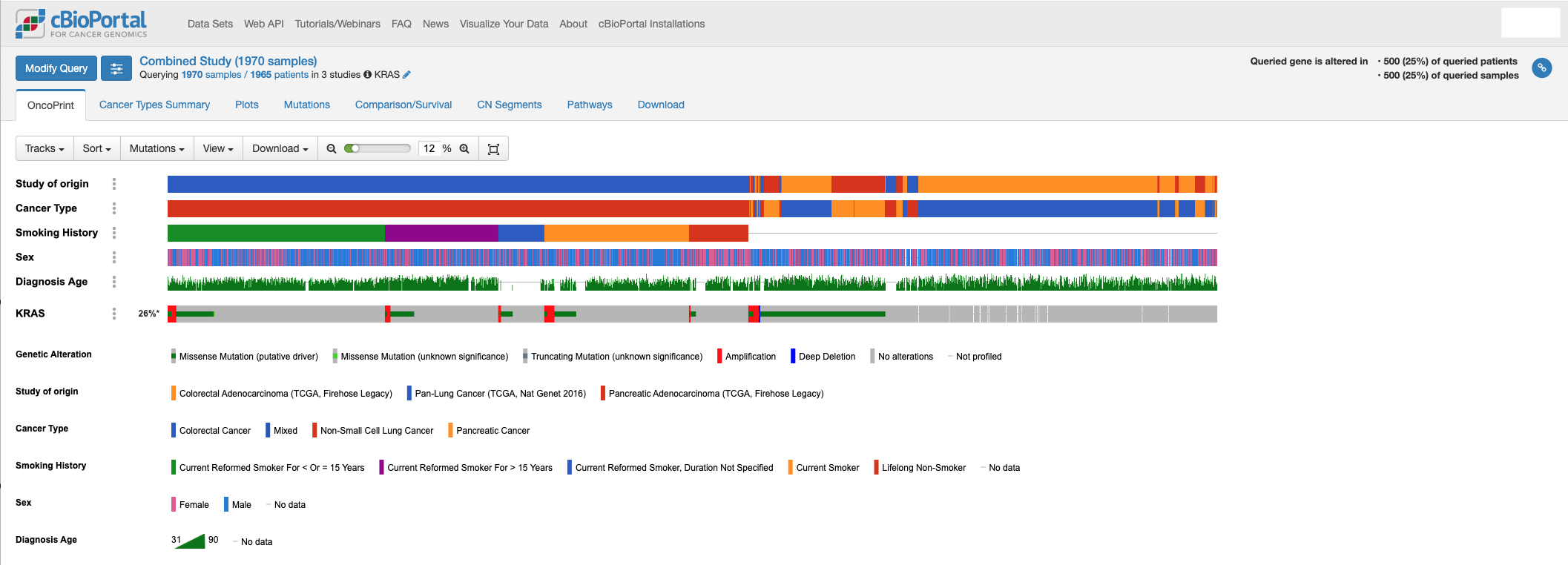
With cBioPortal, you can visualize genomic data, such as mutations, alongside clinical factors like sex, age at diagnosis, or smoking history. This allows for a deeper understanding of how these clinical traits may correlate with specific genetic changes. In the example below, we can add tracks representing patient sex, age at diagnosis, and history of smoking in subjects with mutations in KRAS. You can see that KRAS mutations are frequent in individuals with a history of smoking.
-
You can also correlate if specific clinical covariates are associated with certain mutation types or expression like in the screenshot below. Even if you have a particular tool in mind that you’re more comfortable with, users can download the underlying data and analyze it outside of cBioPortal.
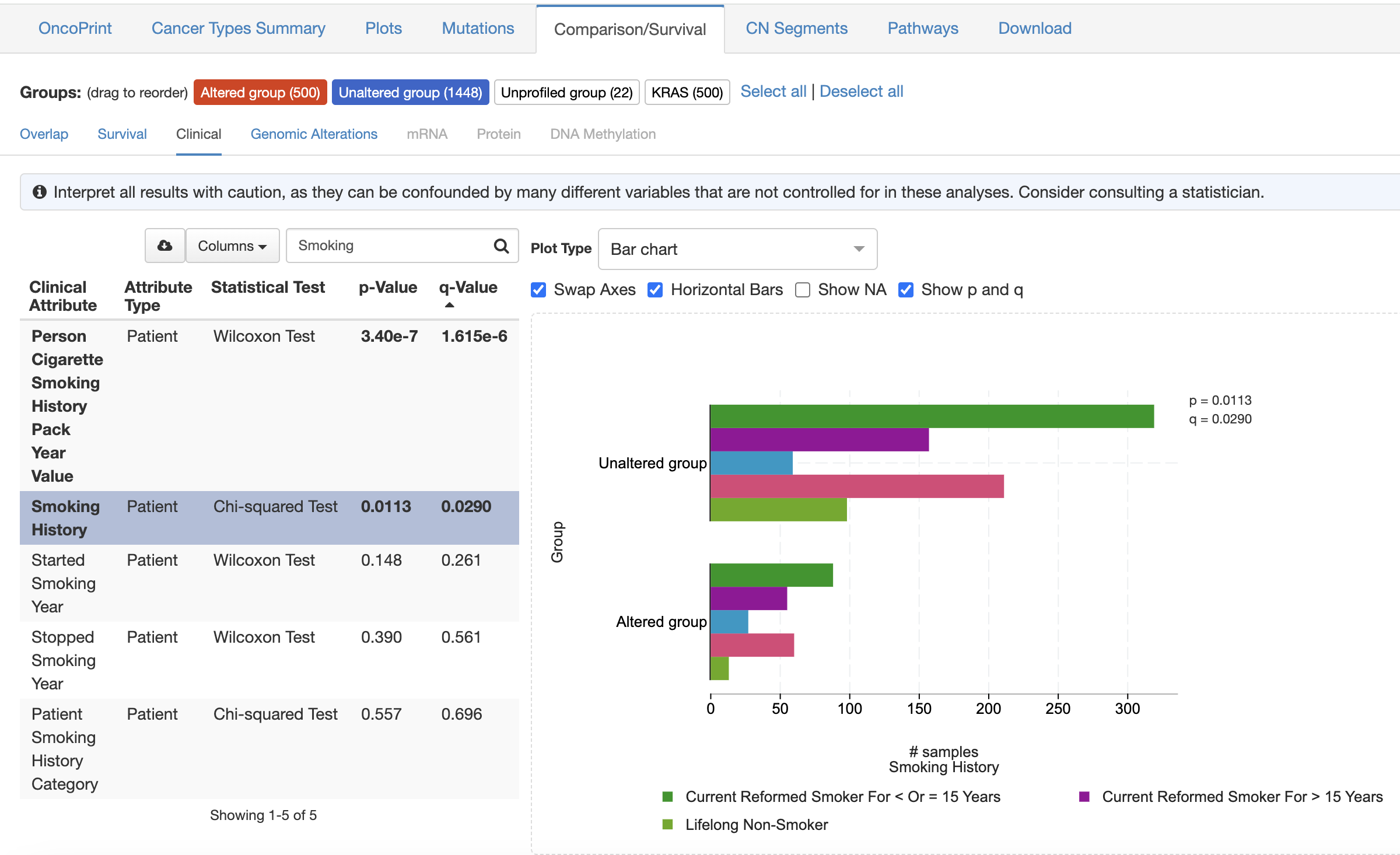
Overlaying clinical data with genomic data is crucial for discovering novel associations between genetic mutations and patient outcomes. This integration can help you identify patterns that may not be apparent from analyzing genetic data alone. By combining both types of data, it becomes easier to uncover critical insights that can lead to better predictions of survival and improved treatment strategies.
Question 3: Do KRAS mutations co-occur with other kinds of mutations?
- You can also utilize the cBioPortal platform to identify if specific genes co-mutate with KRAS. Alternatively, you could assess if they are mutually exclusive to KRAS mutations.
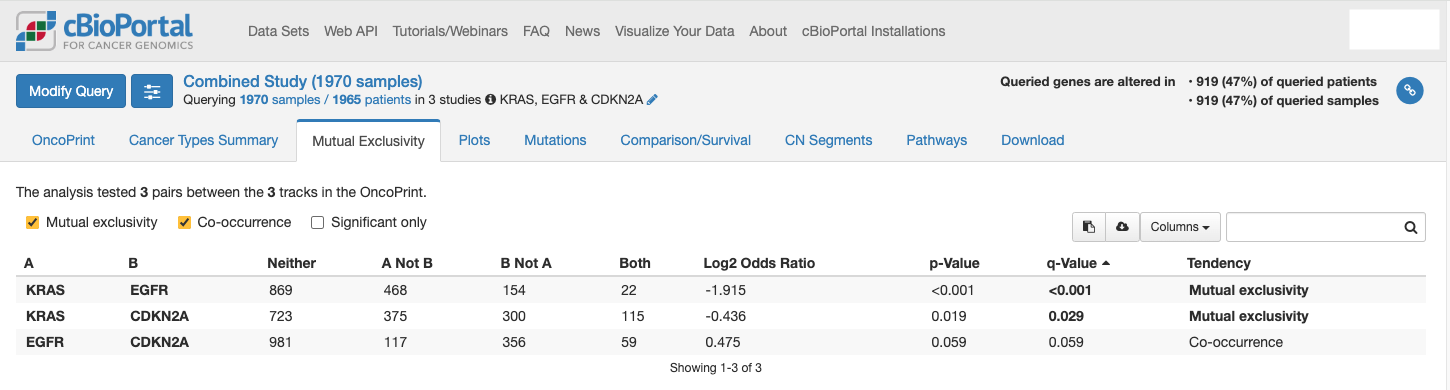
These analyses can reveal alternative therapeutic targets by identifying genes that often co-occur with mutations (co-occurrence) or show mutually exclusive patterns (synthetic lethal interactions). Ultimately, these findings can improve predictions for patient outcomes and guide research direction to identify personalized, effective therapies.
Question 4: Do KRAS mutations affect survival or disease progression?
-
Using cBioportal, you can explore the relationship between KRAS mutations and overall survival in the three cohorts. In the analysis below, it appears the KRAS mutant tumors have a worse overall survival compared to those cancer patients with unaltered KRAS.
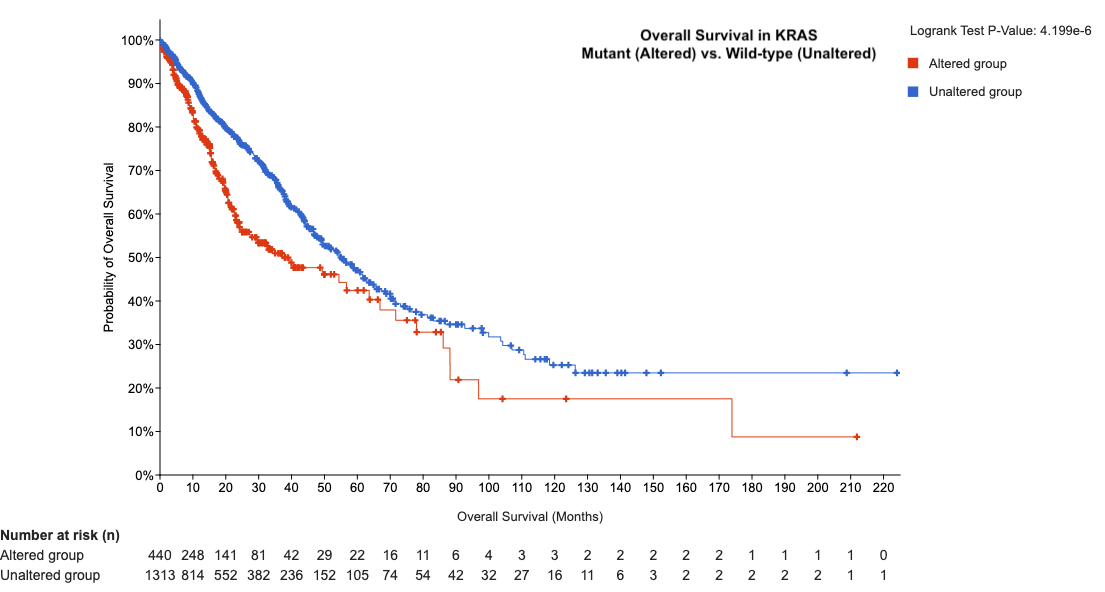
-
You can also explore if KRAS mutations affect disease free survival. In this case, we see KRAS mutant tumors have a lower percentage of disease-free survival which is an important metric when considering oncogenic targets for therapeutic intervention.
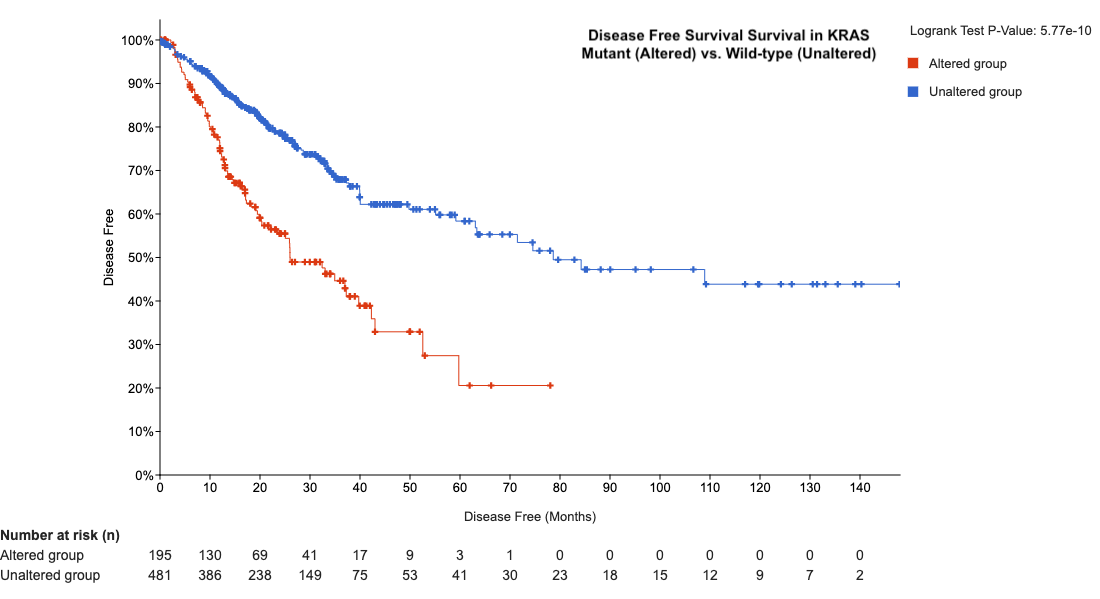
cBioPortal provides a powerful platform for assessing overall survival and disease-free progression by integrating genomic alterations with clinical outcomes. The platform allows you to visualize survival curves, stratify patients by genetic features, and correlate these with clinical variables like treatment response. This helps in identifying potential biomarkers that could predict prognosis or guide personalized treatment strategies, making cBioPortal an invaluable resource for cancer research and clinical decision-making.
Other examples of what you can explore with cBioPortal:
- Pathway-level analysis: Investigate how genetic changes affect key biological pathways using the “Pathways” tab, helping you understand their impact and find potential drug targets. For instance, you could explore how KRAS mutations influence its signaling pathway and look at mutation frequencies in other related genes.
- Copy number alterations: Explore large-scale genomic changes, such as amplifications or deletions, using cBioPortal’s “CN Segments” tab. For example, you could compare copy number profiles of KRAS mutant tumors to non-mutant ones to identify patterns linked to KRAS mutations.
- Create custom cohorts: Build groups based on genetic mutations or clinical data relevant to your research. For example, you could focus on lung adenocarcinoma patients with KRAS mutations and a history of smoking to study genetic and clinical patterns.
- Tumor evolution: Examine how tumors change over time by analyzing multiple samples from the same patient. For instance, you could investigate whether KRAS mutant tumors become more genetically unstable over time, thus potentially affecting survival.
- Immuno-genomics: Study the relationship between genetic changes and immune-related features like tumor mutational burden or immune cell infiltration. For example, you might explore if KRAS mutant tumors have a higher mutational burden, suggesting a more immune-responsive environment.
What is the Fred Hutch instance of cBioPortal?
It is a local installation of cBioPortal within the Fred Hutch computational infrastructure.
- Why this is useful to you as a researcher?
- The Fred Hutch instance of cBioPortal is useful for interim visualization of your clinical and genomic data (i.e., before publication), whether it be for an initial exploration of your dataset, comparison to well-established datasets from outside groups, or just an easy way to make publication-level graphs!
- Hosting your data on the Fred Hutch instance also easily facilitates sharing data (up to individually identifiable research data) with collaborators, provided they have Fred Hutch credentials.
- Study data can be restricted/visible to authorized personnel approved by the study lead/PI.
How can I access the Fred Hutch instance of cBioportal?
- Before accessing the Fred Hutch instance of cBioPortal, you must:
- have Fred Hutch credentials
- be logged in to the Fred Hutch network
- If on-campus, connect to the Marconi network.
- If off-campus, connect via VPN.
- Navigate to cbioportal.fredhutch.org and log in using your Fred Hutch credentials.
How can I request to upload my data into the Fred Hutch instance?
If you are interested in uploading your own data into the Fred Hutch instance of cBioPortal, here are the steps you need to follow:
Note: The cBioPortal team can be contacted via the #cbioportal-support channel on FH-Data Slack, or reach out directly to the DaSL Data Governance Team at dataprotection@fredhutch.org.
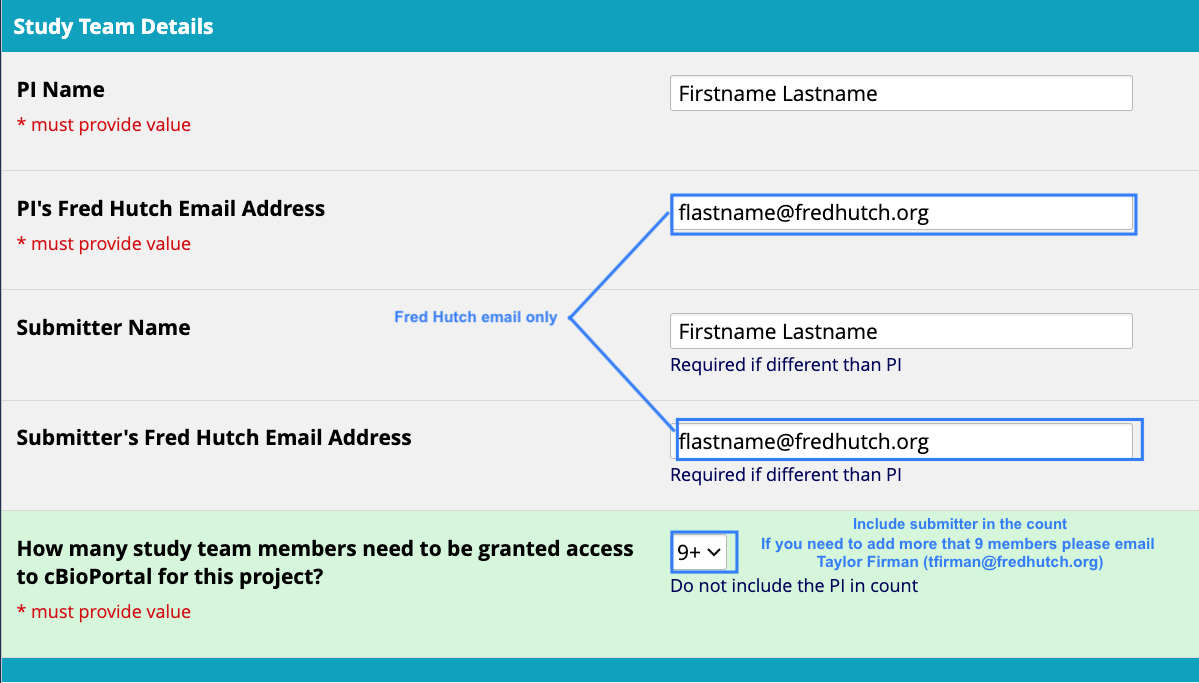
- Request access to upload your study by submitting a response on the cBioPortal Access Request REDCap Form. Before submitting, make sure to prepare the following information.
- Study specific details
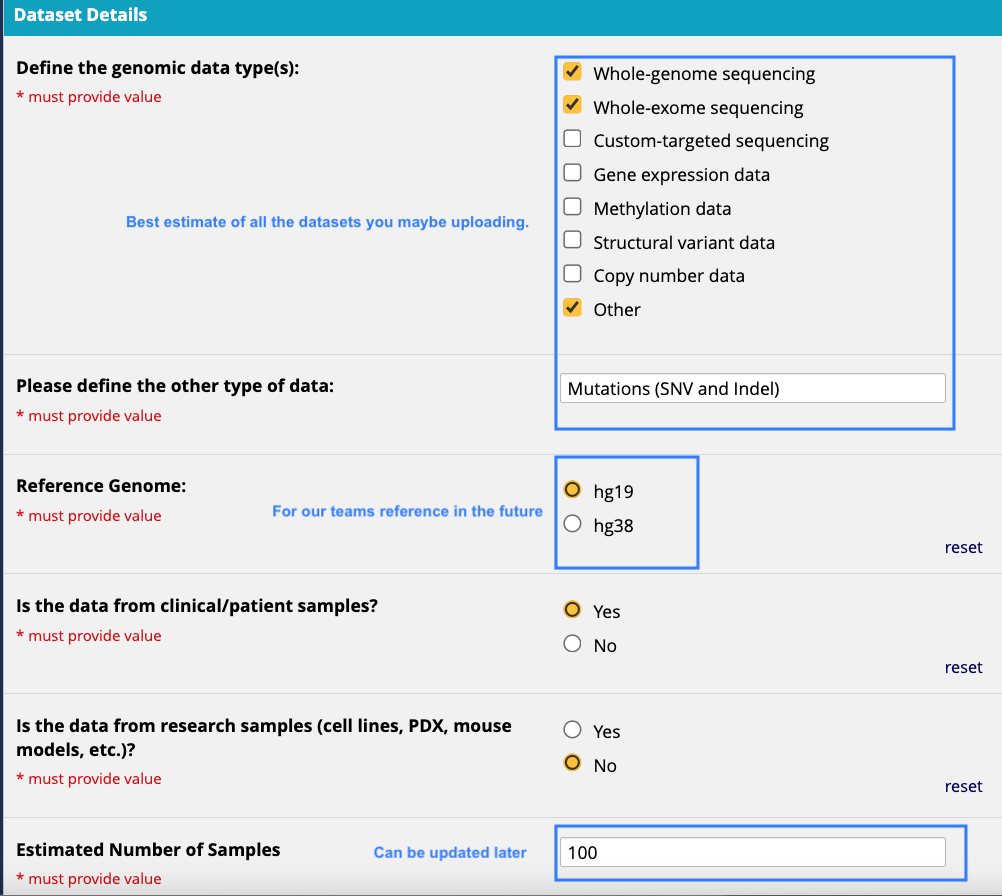
- Will you be uploading PHI data and if so what types?
- What kinds of samples do you plan to upload and approximately how many?
- Who will be processing the study data and uploading it to Fred Hutch cBioPortal?
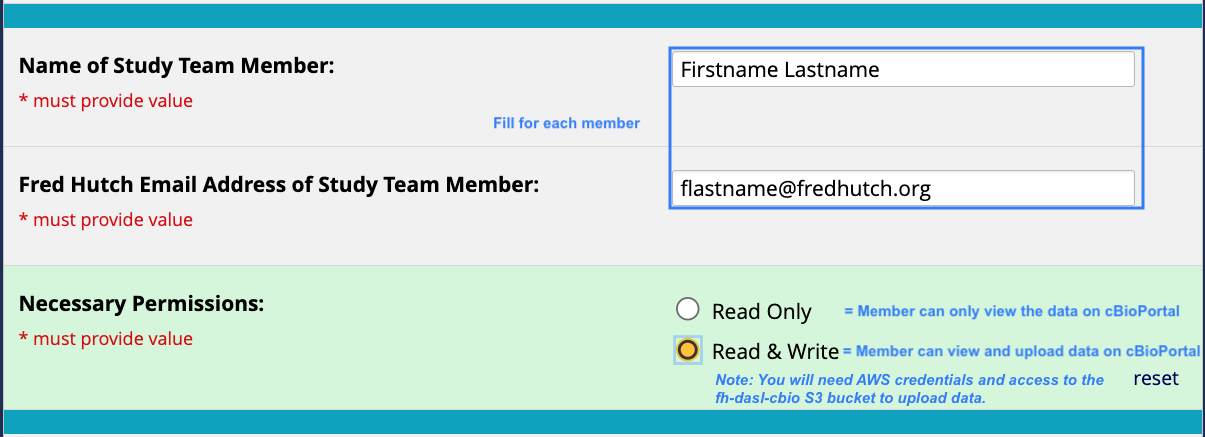
- Which group members should have access to this study upon upload?
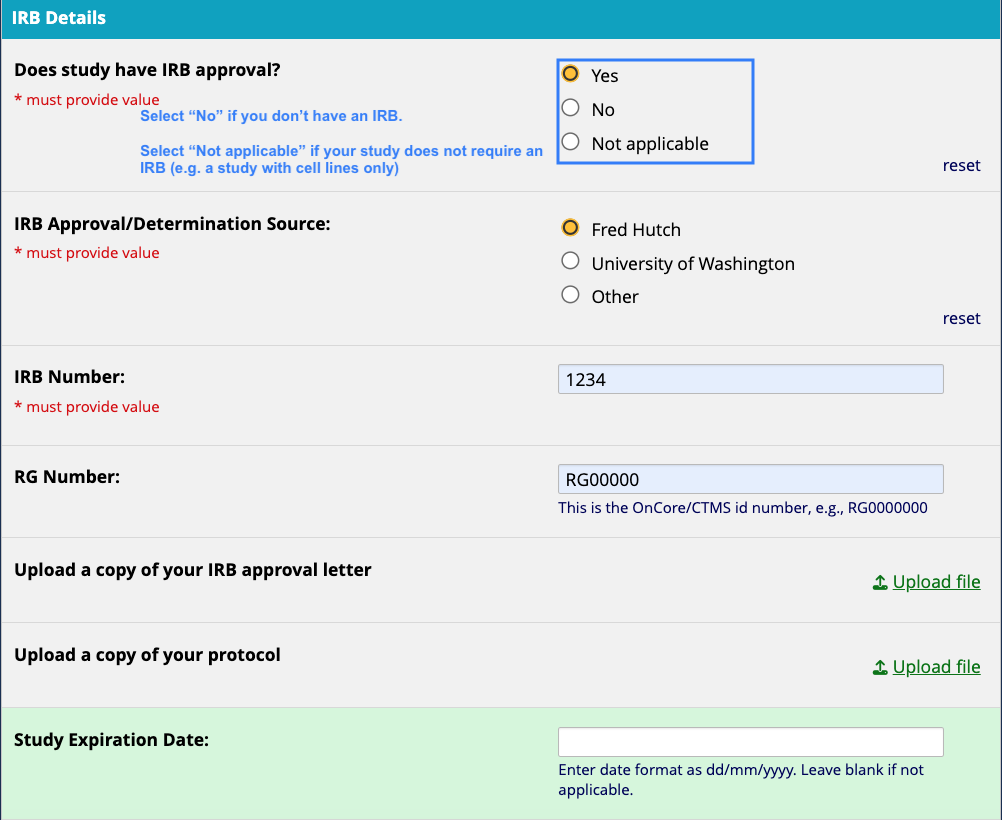
- IRB documentation details
- IRB number
- RG number
- IRB Approval Letter
- A copy of the protocol The cBioPortal admin team will review your submission and notify you via email once it’s been approved for upload.
- Study specific details
Note: Study approval is specific to the IRB and datatypes specified in the REDcap form. While periodic auditing will be performed by the DaSL team, it is the lab’s responsibility to ensure that only the approved datatypes are uploaded to the platform. Failure to do so will result in study removal from the FH cBioPortal platform. If you would like to upload any new datatypes for an existing study, please reach out to the Data Governance team as this will require additional review. If you would like to create a new project covered under a different IRB, please submit another response to the REDCap Form.


-
In the meantime, get AWS credentials by emailing the Fred Hutch help desk. Note: Make sure to include your PI in this email request as they’ll need a lab-based account as well. Once Fred Hutch help desk emails you back with your credentials, make sure to test them to ensure they are functioning correctly. If you already have AWS credentials, skip this step.
-
Get access to the
fh-dasl-cbioS3 bucket by emailing the cBioPortal team your AWS (Amazon Web Services) Account ID number and AWS username. Once the cBioPortal team gives you access, you will receive a confirmation email. Test your access to the cBioPortal S3 bucket by following these steps in a terminal window:# How to test you have the correct access to the fh-dasl-cbio S3 bucket. # Do the following to test if you have the correct access to the fh-dasl-cbio bucket. # You should only be able to write and list files to this S3 bucket. # ssh into rhino and follow the instructions here to configure AWS CLI (https://sciwiki.fredhutch.org/scicomputing/access_credentials/#configure-aws-cli) ssh user@rhino module load awscli aws configure AWS Access Key ID [None]: AKIAIOSFODNN7EXAMPLE AWS Secret Access Key [None]: wJalrXUtnFEMI/K7MDENG/bPxRfiCYEXAMPLEKEY Default region name [None]: us-west-2 Default output format [None]: # You should be able to write a file into the S3 bucket # Write a simple text file into the s3 bucket echo hello | aws s3 cp - s3://fh-dasl-cbio/hello.txt # List aws s3 ls s3://fh-dasl-cbio # You should NOT be able to retrieve/delete any study data (even your own). # aws s3 cp s3://fh-dasl-cbio/hello.txt hello.txt # Should error out... - Prepare your files for upload. This can take some time as cBioPortal requires that your data be in a very specific format (more details on this step below).
- Zip your study folder before moving it into the
fh-dasl-cbioS3 bucket. On a Mac, right-click on your study directory and click “Compress”. On Windows, right-click on your study directory, select “Send to”, then “Compressed (zipped) folder”. If you prefer to use the command line, you can zip the folder using this command:# Go to the directory where your study folder is present cd /path/to/directory/cancer_study_indentifier # Zip the folder recursively zip -r cancer_study_identifier.zip . - Transfer your data onto the
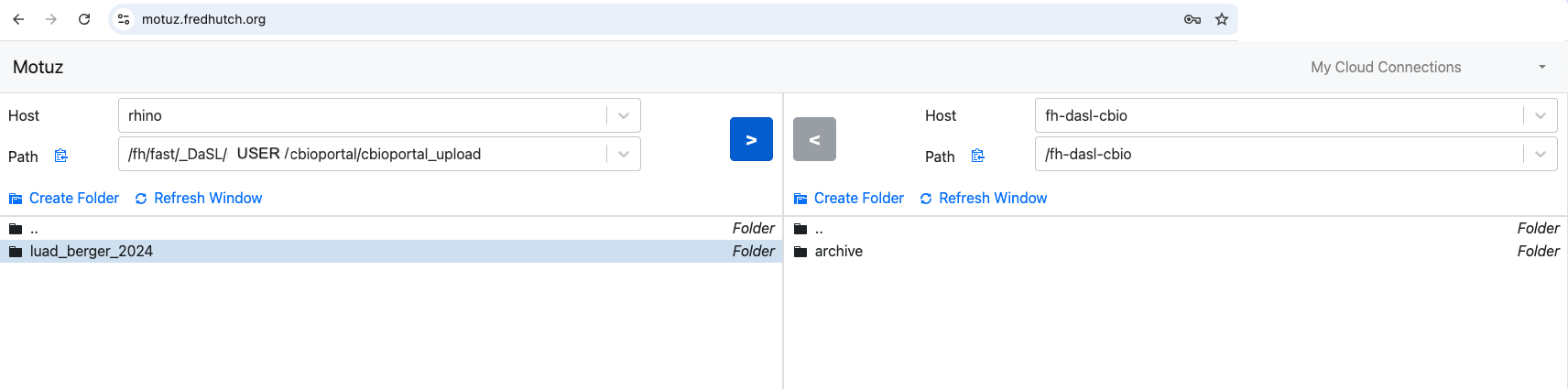
fh-dasl-cbioS3 bucket. You can do that one of these 3 ways:- Motuz:
- Mountain Duck:
- Follow these instructions to setup Mountain Duck.
- Then setup the
fh-dasl-cbioS3 bucket by following these steps. - Open the
fh-dasl-cbiobucket in finder.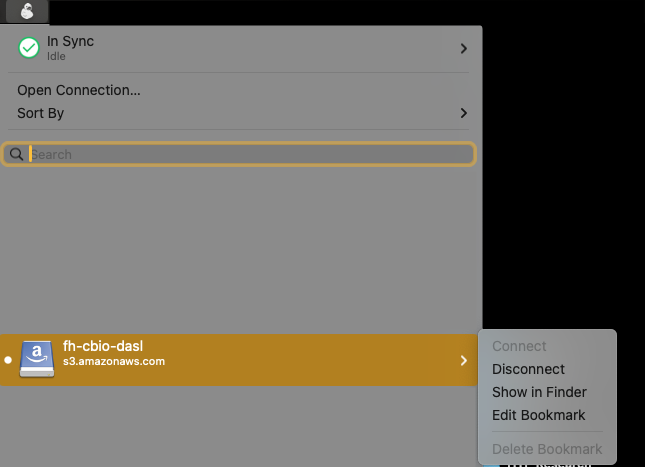
- Copy over your study folder: right-click on the study folder (wherever you have made it), select “Copy”, go to the
fh-dasl-cbiotab or window, right-click and select “Paste”.
- Command line:
Go to terminal and enter the following command to upload your folder into the
fh-dasl-cbioS3 bucket:aws s3 cp /path/to/directory/cancer_study_identifier.zip s3://fh-dasl-cbio/
-
Our Airflow automation scripts will take care of the rest of the upload process from there and send you an email notification with details about the outcome. If upload was unsuccessful, the email will contain a detailed HTML file called a “validation report” that identifies which parts of your study are causing the issue and need updating. Note: If you do not receive an email notification indicating the success/failure of your study upload within an hour, reach out to the cBioPortal Team for help identifying the issue.
-
Go have fun and explore your data on the Fred Hutch instance of cBioPortal!
- If you would like to update your study data or add more subjects to the cohort in the future, simply update your study data locally and reupload a new compressed folder to the
fh-dasl-cbioS3 bucket using the instructions in step 6. Any subsequent uploads of the same study will overwrite the previous study data.
Note: If you would like to upload any of the available public studies into the Fred Hutch instance of cBioPortal, please contact the cBioPortal team via the #cbioportal-support channel on FH-Data Slack.
How do I prepare my data for upload into cBioportal?
Before you begin
- Start by creating a folder to store all of your relevant study files. The name of the folder must be the exact study ID that was provided to you by the cBioPortal team. If any other name is provided, the automation steps will refuse upload since the altered study ID would not be on the list of approved values. The
cancer_study_identifiervalue in your meta files should also be this same study ID. Note: the study ID can be updated if desired, just reach out to the Data Governance team with your preferred ID. - Once your study files are close to complete, you can test out your file formatting using one of two validation methods:
- If you’ve received official approval for upload, try directly uploading it into the
fh-dasl-cbiobucket. As mentioned above, the automated upload process will send you a validation report via email outlining any issues that may have popped up. - You could also validate your study folder by launching a local instance of cBioPortal and troubleshooting the errors (if any) in your study data.
- If you’ve received official approval for upload, try directly uploading it into the
Required/Optional Study Files
There are a few files that are required while all other files are optional. Below is an overview of the required files and some optional files Note: Version 6 of cBioportal currently also requires in the least 1 non-clinical file to be uploaded as well. See instructions below on where to find a dummy table that you can modify in case you are only uploading clinical data.
| Type | Requirement | Filename Example | Required Format | Purpose | Detailed Instructions | Example |
|---|---|---|---|---|---|---|
| Cancer Study | Required | meta_study.txt | Text file | Overall information about the study | Readme | Example |
| Cancer Type | Optional | meta_cancer_type.txt | Text file | A meta file with information about the file with new cancer type. Required if your cancer type does not exist in the database. | Readme | Example |
| Cancer Type | Optional | cancer_type.txt | Tab Separated Value (TSV) | Details about a new cancer type not found in the cBioPortal database. Required if your cancer type does not exist in the database. | Readme | Example |
| Clinical Sample | Required | meta_clinical_sample.txt | Text file | A meta file with information about the clinical samples | Readme | Example |
| Clinical Sample | Required | data_clinical_sample.txt | Tab Separated Value (TSV) | File with the sample-level clinical covariates/metadata | Readme | Example |
| Clinical Patient | Optional | meta_clinical_patient.txt | Multi-line text file | A meta file with information about the clinical patient | Readme | Example |
| Clinical Patient | Optional | data_clinical_patient.txt | Tab Separated Value (TSV) | File with the sample-level clinical covariates/metadata | Readme | Example |
| Panel | Optional | meta_gene_panel_matrix.txt | Multi-line text file | A meta file for describing the gene panel matrix file | Readme | Example |
| Panel | Optional | data_gene_panel_matrix.txt | Tab Separated Value (TSV) | Sample level details of the gene panel used for the different samples | Readme | Example |
| Mutation | Optional | meta_mutations.txt | Multi-line text file | A meta file describing information about the mutation file. | Readme | Example |
| Mutation | Optional | data_mutations.txt | Tab Separated Value (TSV) | File with mutation data | Readme | Example |
| Case Lists | Required | case_lists/cases_sequenced.txt | Multi-line text file | It helps cBioPortal identify which samples have data. Required if uploading data files beyond clinical data. | Readme | Example |
| Structural Variant | Optional | meta_sv.txt | Multi-line text file | A meta file for describing the structural variant data file | Readme | Example |
| Structural Variants | Optional | data_sv.txt | Tab Separated Value (TSV) | File with structural variant data | Readme | Example |
| Generic Assays: Arm-level CNA | Optional | meta_armlevel_CNA.txt | Multi-line text file | A meta file for arm-level copy number alteration data | Readme | Example |
| Generic Assays: Arm-level CNA | Optional | data_armlevel_CNA.txt | Tab Separated Value (TSV) | Arm-level copy number alteration data | Readme | Example |
Publicly available tools for data formatting
There are many publicly available formatting tools that may help with the formatting process. When deciding which one works best for you, it ultimately depends on what tools you’re comfortable with and what kinds of data you’re uploading, but here are a few options that might help get you started:
| Tool Name | Description | Advantages | Disadvantages | Fred Hutch Repository Link |
|---|---|---|---|---|
| Data-processor | Formats clinical data tables in multi-tab Excel files to cBioportal format | - Useful for varied clinical data fields. - Supports multi-tab Excel files. - Easy terminal execution |
- Does not seem to work to generate clinical data files. - Requires adherence to specific clinical data variable names from [cBioPortal Clinical Data Dictionary] |
Data_processor |
| cbpManager | An R-based Shiny App that allows users to create and upload cBioPortal-formatted studies. | - A relatively easy to run R based (Shiny) App. - Allows you to create clinical data files, timeline related files, and mutation files. - Allows users to run the validation of their formatted study folders |
- Currently only helps to create clinical and mutation data related files. - If using the app to create the files then can only update one patient at a time |
cbpManager |
| CaisisTools (a Fred Hutch tool) | Takes clinical data in the form of an excel workbook and converts to cBioportal format | - Helpful for processing clinical data. - Can be used for data from RedCap |
- Data either must be obtained from Caisis or should be in the same format | CaisisTools |
| Varan 2.0 | Takes genomic data and existing study folder to process and upload into cBioportal. | - Useful for validating an existing cBioportal study folder. - Can concatenate from multi-sample vcf files. - Can be used to do filtering of genomic files |
- Has several local dependencies (vcf2maf, VEP, and samtools). - Folder preparations restricted to CNV, SNV, SV, and clinical data |
Varan |
| cBioPortal-BS-Lab | Helpful scripts to take data from RedCap to convert to clinical data files | - Good for demonstrating how to take data stored in RedCap and format | - Mostly would be useful for clinical data files | cBioPortal-BS-Lab |
| cBioPortal_Importer | Python script to prepare data for uploads into cBioportal. Mostly genomics data. | - Helpful scripts to transform specific data types into cBioportal format | - Accepts very specific output files. - Requires threshold setting for copy number data, etc. |
cBioPortal_Importer |
| cbpConverter | R Shiny App to convert Excel sheets into cBioportal format | - Seems like a simple Shiny app to convert clinical data into cBioportal format | - Looks untested but might have helpful scripts. - Again only clinical data |
cbpConverter |
| gdc-et-pipeline | Converts data from the GDC repository to cBioportal format | - If data is available on GDC, this might be useful | - Written in Java. - File formats have to be in the GDC format. - Folder preparations restricted to CNV, SNV, Expression, and clinical data |
gdc-et-pipeline |
| kf-cbioportal-etl | Specific to this study: CAVATICA and Data Warehouse | - Helpful scripts that can be leveraged. | - These scripts might be specific to the format of files found in this study. | kf-cbioportal-etl |
| mutational-signature-converter | Very specifically converts the mutational signature data into cBioportal format | - Helps to convert mutational signature data into cBioPortal format | - Has not been updated in a few years. - Simple python script |
mutational-signature-converter |
| shah-cbioportal-tools | Specifically for formatting Copy Number Data expects a seg file and TITAN output | - Could potentially be used for tools other than TITAN that generate a seg file | - Specific for TITAN outputs | shah-cbioportal-tools |
Quick Links
- Fred Hutch cBioPortal Demo Day Recording
- Public instance of cBioPortal
- cBioPortal data formatting instructions
- Fred Hutch data formatting tips and tools
- Publicly available cBioPortal datasets
- Video tutorials on using cBioPortal
Help
Report bugs or issues
To report bugs or issues with the Fred Hutch instance of cBioPortal, please file an issue here. For questions about using the tool and formatting your data for upload, schedule a data house call using the link above.